
Note: This piece was published as a Commentary in the journal Structure! You can find it (paywalled) here, but I’m allowed to share the submitted version open access on my blog! Hope you enjoy, it’s one of my favorite things I’ve written to date.
During global pandemics, the spread of information needs to be faster than the spread of the virus in order to ensure the health and safety of human populations worldwide. In our current crisis, the demand for SARS-CoV-2 drugs and vaccines highlights the importance of biological targets and their three-dimensional shape. In particular, structural biology as a field was poised to quickly respond to crises due to previous experience and expertise, and because of its early adoption of open access practices.
Keywords: SARS-CoV-2; structural biology; open access; Protein Data Bank; Electron Microscopy Data Bank; Electron Microscopy Public Image Archive; validation metrics.
While many research laboratories were forced to shut down due to the SARS-CoV-2 (CoV-2) pandemic, scientists have been working hard, albeit six feet apart, to find out everything they can about the virus. Necessary changes due to the pandemic highlight how and where the scientific process both succeeds and needs improvement. Structural biology quickly adapted to studying the inner-workings of the CoV-2 virus.
Finding a drug to help CoV-2 patients or a vaccine to prevent the spread has been given top priority as drug candidates rush through clinical trials. The bio-pharmaceutical industry relies on structural biology because 3D structures provide information about potential binding pockets and show how small-molecules bind to their target(s) to elicit a biological response. Thus far, CoV-2 protein structures determined using two techniques, protein crystallography and cryo-electron microscopy (cryoEM), have primarily been used to determine experimental models (Fig. 1A).
As a field, structural biology was poised to respond to a biological emergency. Over the past decade, updates and adaptations to techniques and methods have made structure determination a relatively quick process that can swiftly shift focuses and goals. In addition, the field heavily relies on open access databases and resources which allow for accessible and interoperable use of structures for research and education.
Right on Target: Structures for drug and vaccine design
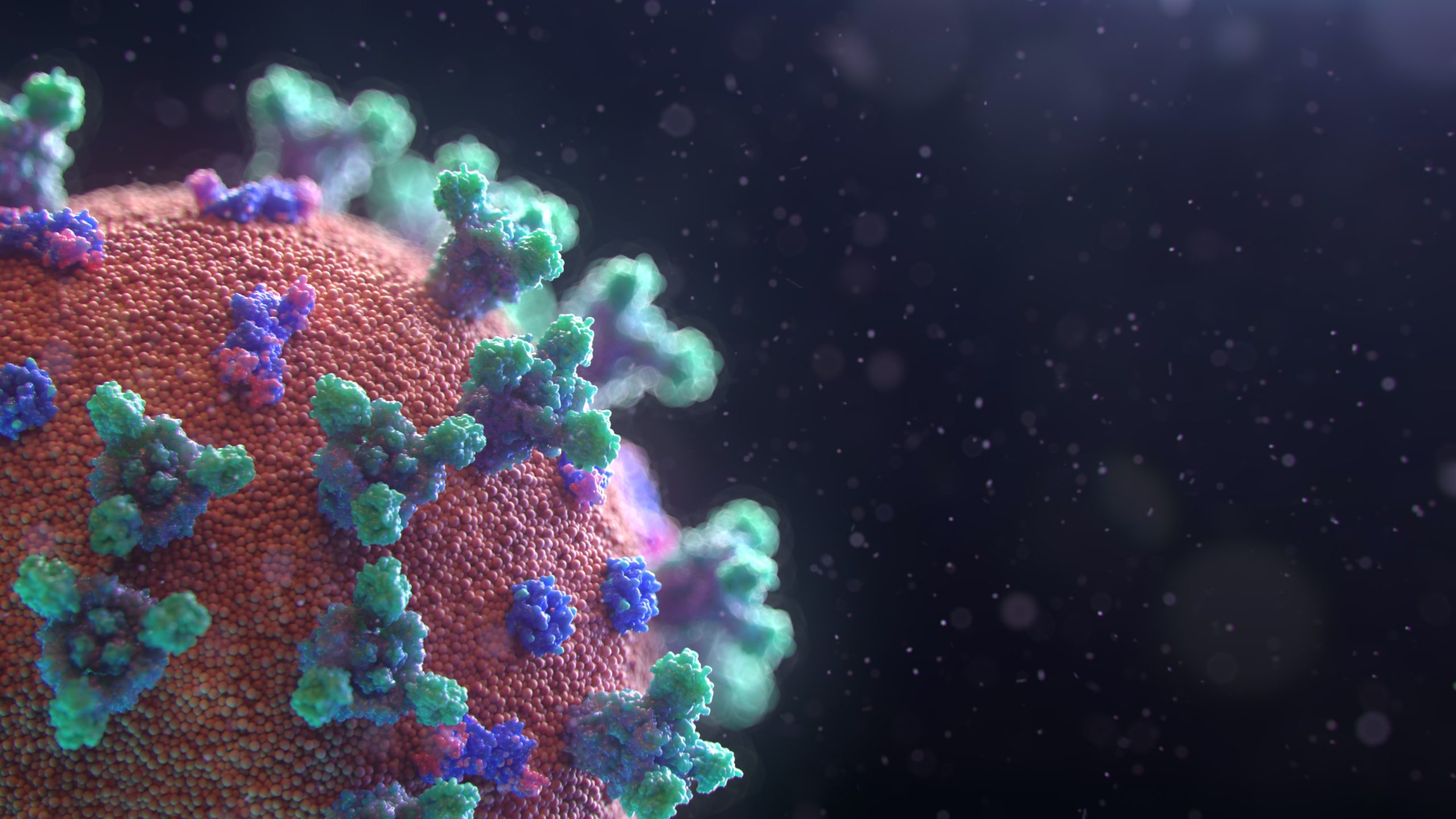
Those who are either lucky or brave enough to remain in the lab, have been in an accelerated business-as-usual. “There’s a sense of urgency that we normally don’t have,” says Daniel Wrapp, a graduate student in the McLellan lab. Wrapp, along with postdoctoral fellow Nianshuang Wang, co-authored an open access publication describing structures of the CoV-2 spike glycoprotein in early-March (Wrapp et al., 2020) (Fig. 1C). Formed in groups of three, spike proteins arrange themselves on the surface of the virus. This crown-like architecture of spikes recognizes proteins called receptors on the host cell surface which allows the CoV-2 to infect. Spike proteins recognize host receptors, specifically ACE2, when one of the three spikes is in an “up,” conformation (Walls et al., 2020). The region that CoV-2 binds to the receptor is of particular interest for drug and vaccine development.

A) Number of CoV-2 structures deposited and released in the Protein Data Bank (PDB) as of 7/8/20, plotted by experimental method. Colored bands are ordered by date deposited and correspond to B) the first CoV-2 structure determined, the main protease (red, pdb 6lu7), C) CoV-2 spike protein (yellow, pdb 6vsb), D) an RNA-dependent RNA polymerase (green, pdb 7btf), asterisk shows the region discussed in Fig 2AB, and E) the receptor binding domain with an antibody (blue, pdb 6w41).
Conveniently, CoV-2 spike protein is comparable to the SARS spike, sharing 76% protein sequence identity (Ou et al., 2020). As such, many labs have been able to jump-start CoV-2 projects based on their previous experience with coronaviruses. The similarity between SARS and CoV-2 spike proteins in function and sequence allowed Wrapp and Wang to “go from cloning, to expression and purification, to grid preparation within just a week. If this was the first time [they] had expressed any protein in this sort of family, there’s no way it would have been that quick.”
Even labs that don’t work on coronaviruses have expertise that applies to CoV-2. Take Dr. Ian Wilson, a principal investigator at Scripps, who has been studying viral pathogens and vaccine design for viruses, like influenza and HIV. Even though CoV-2 falls into a different family of viruses than either the flu or HIV, they are all enveloped viruses and use a similar mechanism to invade cells and evade the cellular immune system. The spike proteins on the surface of CoV-2 are covered in sugar-protein complexes called glycoproteins that make it difficult for the host cell to recognize CoV-2 as a threat. Glycoproteins at the surface of viruses also help identify receptors that allow the virus to bind and invade the host cell. Researchers have been studying the mechanism of viral invasion and the role of their surface glycoproteins, Wilson himself identifying the first glycoprotein of the influenza virus (Wilson et al. 1981). Since then, his lab has determined many structures of enzymes with neutralizing antibodies, and are now looking at antibodies to the CoV-2 receptor binding domains of the spike protein (Yuan et al., 2020) (Fig. 1E).
A big part of a researchers’ ability to develop small molecule anti-viral drugs or antibodies relates to knowledge of what protein targets look like from using structural biology tools. “In a sense, we were very prepared to work on CoV-2 because of the many years developing tools and necessary state-of-the-art equipment for producing viral antigens, characterizing them, and imaging them with their intended target,” says Wilson. Some of the glycoproteins can be tricky to produce in large quantities and stable enough to see using crystallography or cryoEM, “but if you get a structure, you have a visual indication that you’ve done it right.” Seeing is believing, and experimental models showcase interesting biological features, in this case, to identify compounds that could prevent or treat CoV-2.
It’s fortuitous that many CoV-2 features and structures follow a similar pattern or shape as previous coronaviruses like SARS. “Of course, not all Covid proteins behave, but if you look across the proteins we’ve been able to express so far we are doing pretty well, which is unusual,” says Dr. Karolina Michalska, a researcher at the Advanced Photon Source (APS). “The overall success rate for structural genomics is probably around 5%, and I think we are doing much better. It’s one lucky moment in the whole pandemic situation.” At the APS, Dr. Michalska is a protein crystallographer and has been involved in determining many crystal structures of proteins related to CoV-2. Light sources, such as the APS, are essential for this work. Access to beamlines for CoV-related projects is limitless given their high priority, and the established organizational and experimental set-up of the APS itself enables rapid structure determination. The same can be said for the newer cryoEM facilities and national centers, where microscope time is given to CoV-2 related projects. In addition, the APS focused intensely on SARS-related proteins during the outbreak in 2003, so they have protocols ready to apply to study CoV-2.
Tracing back the biochemistry, experimental work on CoV-2 proteins originates from the rapid sequencing of the first CoV-2 genome back in late January 2020. The adoption of sequencing data to structural work, along with bioinformatic analysis to understand virus evolution, is only possible because the sequence was made freely and openly available. Easy access to the most recent data and information is critical, as researchers world-wide work together in tandem to understand and seek treatments and vaccines for the virus.
Open for business: history of accessibility
Sharing the methods going in and data or publications coming out of research has become central to researchers who are globally uniting to tackle the pandemic as an integrated front. Open access (OA) as an infrastructure and philosophy makes research output freely accessible and discoverable. OA increases availability of information and scholarly communications for reading, sharing, and reusing information. The practice of open access broadens the visibility of research, increases the impact of scholarship, and enriches the public’s knowledge for informed decision making (McKiernan et al., 2016).
The practicality and advantage of easing the spread of information, be it in the form of raw data or a scholarly publication, has not gone unnoticed. Typically, the barriers to publications in the form of paywalls limits access to the most up-to-date peer reviewed research. Many paywalled journals in the wake of a pandemic are waking up to researchers needing immediate access to data and publications. By making all coronavirus-related articles discoverable, accessible, and reusable the goal is to spread data to stop the spread of the virus.
For structural biology, the Protein Data Bank (PDB) acts as a preservation steward of structural data following OA FAIR principles (Wilkinson et al., 2016). Established in 1971, just two years after the internet, the PDB is the first open access digital data resource among all biology and medicine resources. Right when it first was created, the PDB stored atomic coordinates of twelve structures, with the coordinates themselves punched in 80 characters per line on physical cards. The spirit of transparency, trust, and availability have continued as online and digital technologies advanced through the present day, so now anyone can analyze and view structures instantaneously through a web browser. The PDB focuses on biocuration, archive management, data exploration, and outreach, which allows for structural biology data to impact basic and applied research in a wide variety of applications (Burley et al., 2018, 2019). As a database that does more than store data and additionally annotates content, the PDB, along with partner databases like the Electron Microscopy DataBase (EMDB) and Electron Microscopy Public Image Archive (EMPIAR), provides a priceless resource to not only the field of structural biology, but to scientific research as a whole. “The PDB is obviously an essential resource. They develop various tools to make structural research, especially Covid-specific, more accessible and so easy to file. It’s definitely necessary for our work to disseminate our results,” says Michalska.
Typically, structures do not need to be deposited into a database until publication, but “it often takes much longer to write a paper than to produce a structure,” states Michalska. While papers and peer review about CoV-2-related research are also given top priority, often structures deposited into the databases are being released to the public before publication. At the PDB, biocurator Brian Hudson describes that they are keeping track of CoV-2 structures by giving them top priority, “making sure things are moving in a timely manner so people can see and access the structures.” In this special case during an international emergency, “delay can be significant in terms of lives and we don’t know which of these structures is going to be the truly important one” in terms of drug or vaccine targets.
Trust, but validate
Within the context of structural biology, a significant value comes from the expert validation, along with biocuration of the model, structure factors, and atom coordinates during structure deposition into the PDB. Validation provides objective assessments of structure quality, and biocurators validate incoming structures using established parameters and software. Biocurators work to verify or correct structure coordinates, confirm chemical consistency of the 3D coordinates and any bound ligands, and in general make sure that the deposited model checks with the experimental data (Burkhardt et al. 2006).
There are many challenges along the way coming from information validation. While communities have been flattening curves of virus spread, the amount of new data being shared and distributed about CoV-2 is anecdotally exponential. With many studies and data being published, some on pre-print non-peer reviewed servers like bioRxiv, it is difficult to double-check that all the information coming out is reliable and dependable. Despite the careful measures taken by researchers in determining and solving structures, PDB biocurators, and validation metrics, errors still exist. While deposited structures generally haven’t seen a decline in validation scores during the pandemic, some scientists worry that some of the deposited structures are not accurate. “These things are coming out awful fast and there could be a conflict between getting it right, but getting it fast, too,” says Hudson.
In some cases, there is real cause for concern. When Dr. Tristan Croll, a researcher at the University of Cambridge, started to look at the then recently released structure of an CoV-2 RNA polymerase (Fig. 1D), he realized that some of the model did not fit into the electron density map. Using a molecular dynamics-based software, ISOLDE (Croll, 2018), he was able to find that a 31-residue stretch model was shifted out of register by nine residues (Fig. 2A-B). Doing some more investigative research, he proposed that this shift in amino acid registry in the CoV-2 structure likely derived from a similar issue in the SARS equivalent (pdb 6nur). Since then, the atomic coordinates have been updated under the original ID and have been used for atomic model building (Hillen et al., 2020). Croll certainly doesn’t blame researchers for trusting their predecessors, but “these types of issues arise when they’re too much in a hurry to go through and check the structure thoroughly themselves.” Especially with cryoEM datasets, “choosing to refine against certain metrics, like Ramachandran and rotomer restraints, can hide mistakes, rather than fix them,” Croll warns. This results in a model that checks all the boxes for validation metrics, but might still be incorrect at first like the CoV-2 and SARS RNA polymerase.

Even though model validation is tricky, the structural biology community constantly discusses and updates validation statistics to keep up with the rapid changes to the field in terms of new technologies and software. Method-specific validation task forces determine which experimental data and metadata from data collection should be archived and how these data and the derived models should be verified. For example, since 2012, an annual Electron Microscopy Validation Task Force Meeting concludes with recommendations to increase the impact of electron microscopy in biology and medicine (Henderson et al., 2012). Periodic conversations between experts in the structure field regarding validation statistics create a culture that supports best practices in data curation and accessibility. Combining a myriad of knowledge from software developers, wet lab researchers, and data curators combines perspectives and expertise to drive the field of structural biology in useful directions.
As standards are approved by the community, they become integrated with structure databases. In addition, the PDB re-runs validation reports so all structures deposited are standardized and consistent. To further ensure consistency and quality of the archive, the PDB periodically improves or remediates structural data. “The way people were thinking about structures back in 1974 is not the same way that people are thinking about structures now, so the representation file formats have to evolve over time.” Christine Zardecki, the Deputy Director of Education and outreach at the PDB, explains. For example, the PDB had standardized nomenclature and structural depiction of viruses and large biological assemblies (Lawson et al., 2008) and is currently remediating carbohydrate data. “Changing the representation makes the data more organized, consistent, and readable.” These updates are important because of the accessibility of the PDB. “Anyone can check for themselves structure quality,” describes Michalska, “but the average user or consumer who looks into the details might not understand everything that’s available. That’s also why the PDB is important, to educate the end users about the validation tools available.”
A model for scientific research
Structural biology is only a part of any biological story, but a critical and visual narrative component. The 3D shape of a protein tells a lot about it’s activity in a cellular context, and as such drives researchers’ understanding of biological mechanisms and serves as a means to identify target areas for drug and vaccine design. Now more than ever, the pandemic has highlighted how structural biology adapts quickly to respond to real world problems. “Structural biology is nimble and versatile,” says Wilson. “It’s well positioned to integrate into a bigger context and can answer quite complex problems.”
The field of structural biology is not only poised to respond to biological emergencies, but in a bigger sense serves as a model for how research at large can operate, during an international crisis or otherwise. This versatility and nimbleness comes from the practices the structural biology community has adopted including open access to models, attention to changing validation metrics, and the ways that the community integrates with other fields.
Built into the practice is sharing the experimental model along with the methods and validation metrics that go along with it. Open and accessible databases create an environment that’s “ready for ingenuity and creativity in solving problems,” Hudson describes. As a pioneer for OA, the PDB has always insisted on freely available data. Moreover, facilities like the APS and cryoEM national centers additionally increase the accessibility to collect data, with more centers cropping up around the world. While many fields and organizations are realizing the usefulness of OA, there are “barriers to getting data deposited and stored, and adopting the open mindset can be challenging” when infrastructural changes are being made, describes Zardecki. “Structural biology has an advantage, in that sense, to answer questions faster” because the practice is already established.
Given the attention and mindfulness in where the field can improve, the ingenuity is applied and discussed outside of the validation task force meetings. The structural biology community has largely adopted the practice of publishing pre-prints and passionately discusses them on social media platforms. Indeed, Croll went to Twitter to first share his findings that some of the CoV-2 structures looked strange, and distribute his re-modeling process. “Scholarly communication has been designed to be so impersonal: the nice thing about Twitter is that people get to be human and can talk about challenges and ideas more openly.”
Additionally, it seems structural biologists are willing to adapt to community guidelines, even when they are not mandatory. Being aware that mistakes happen in the rush to go from cryoEM models to maps, a call was made to the community to deposit raw data to EMPIAR. By sharing raw data, there could be a global effort to re-process and re-analyze these data to make sure structures are accurate before biologists and bioinformaticians use them, like the recently deposited data of CoV-2 virions (Turoňová et al., 2020). Wrapp “really likes the idea of depositing raw data and using it for crowd-sourcing, because the more eyes on something the better.” Increased involvement in data processing and map to model validation, like what Croll was doing on Twitter, ensures the quality of data being used by so many researchers studying CoV-2 as a transparent and accessible form of peer-review.
As a visual field, structural biology allows anyone to have “the ability to visualize complex biological systems,” Hudson says. “There’s a lot of science that can be very abstract, but here you have something you can hold in your hand and examine. It’s like playing with Legos, where you can see the reason pieces interact and fit together.” Structural biology is “engaging for anyone to play with, not necessarily just professional scientists,” Croll echoes. Given the visibility structures have in the public media, there’s an important role that structures can play in outreach and education especially since databases are available to anyone with internet access.
On the horizons of structural biology, beyond CoV-2 protein structure determinations, the field continues to adopt new methods, tools, and technology. As the world begins to open up again, research labs and institutions could use the opportunity to restart research implementing open access principles, leveraging community engagement and democratic processes to enhance their research and impact.

Genomic (grey) and protein sequences (yellow) are deposited into open access repositories. Once a structure is solved, like the spike protein displayed, it’s experimental data, map, and model are also deposited. Members of the community can then (re)process the data or validate the model (green), or could find an error and update the model (pink). Structures also inform drug and vaccine development, and are used for education and outreach purposes (blue), highlighting the diverse communities that can give back and contribute to structural biology research.
Acknowledgements
I would like to thank everyone interviewed for their work and their time meeting with me; Kevin Boehnke, Jimmy Brancho, Elyse Petrunak, Aidan Sawyer, Sara Talpos, Alex Taylor, and Liz Wason for interesting and constructive conversations; and Kristen Verhey and Michael Cianfrocco for their mentorship, support, and encouragement.
Leave a Reply